Recent Publications
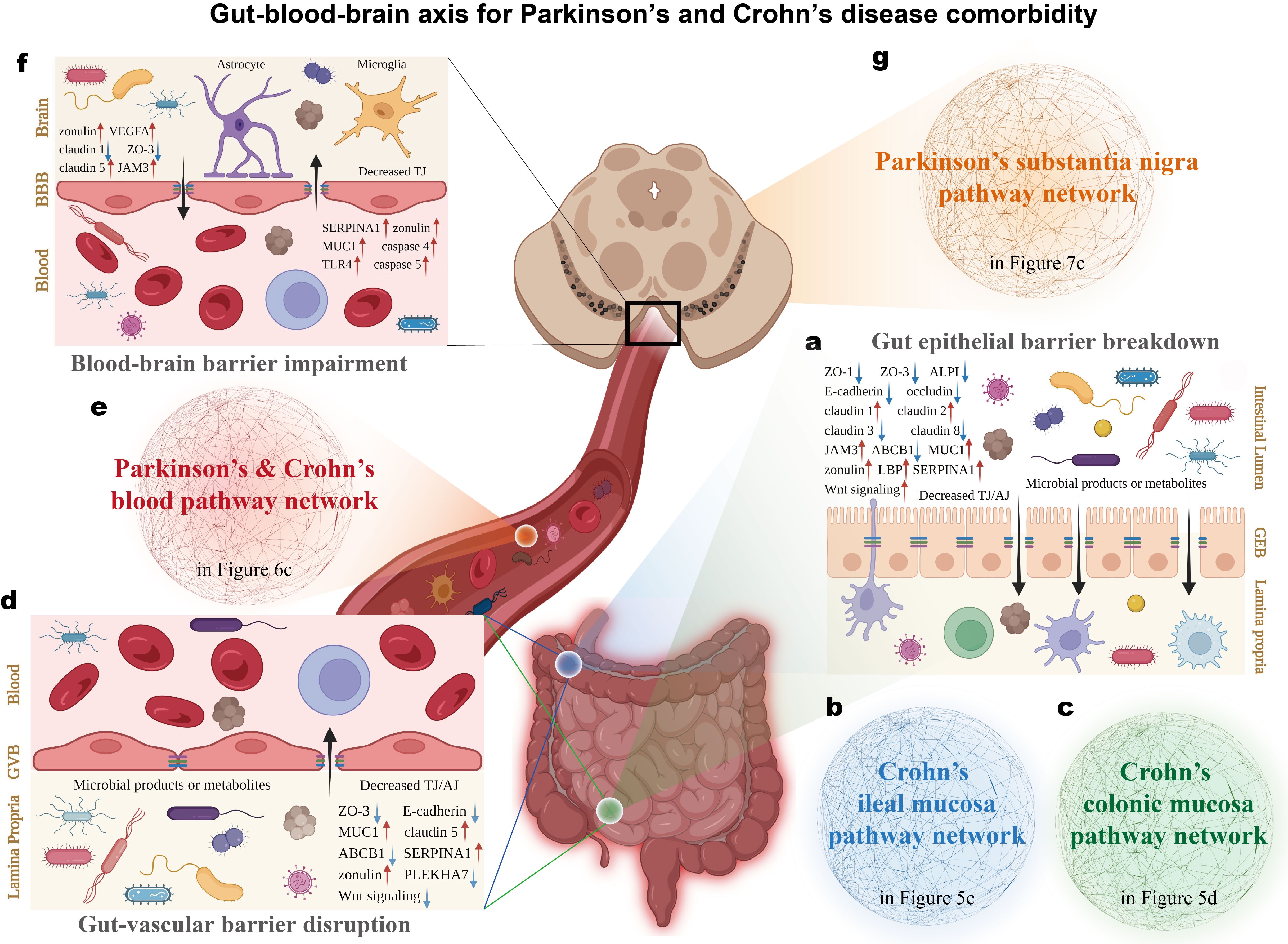
Mapping the comorbid landscape of Parkinson's disease and Crohn's disease along the gut-blood-brain axis
Parkinson's disease (PD) and Crohn's disease (CD) are primarily localized to the brain and gut, respectively. Nevertheless, epidemiological evidence increasingly links these two seemingly unrelated disorders. Although genomic or transcriptomic efforts have been dedicated to understanding this phenomenon, the precise landscape underlying this comorbidity remains elusive. Here, a systematic multi-omics approach is employed to panoramically map this pathogenic nexus for the first time. By curating a comprehensive genetic corpus related to PD and CD from extensive publications, we uncovered a shared genetic architecture converging on biological functions governing host-pathogen interactions and barrier integrity maintenance. Further, multi-tissue transcriptomic datasets were meta-analyzed to validate genomic insights in transcriptional circumstances, which identified pervasive transcriptional synergies of PD and CD pathways within the blood context, indicating in blood CD pathological milieu could create a permissive environment for PD pathogenesis. Finally, delineating the aberrant gut-blood-brain axis through the sequential compromise of gut epithelial barrier, gut-vascular barrier and blood-brain barrier, we revealed a directional cascade where CD intestinal pathology facilitates PD substantia nigra degeneration via blood circulation, establishing a theoretical foundation for preventive and therapeutic interventions for PD and CD comorbidity. Crucially, this study provides a blueprint for dissecting the molecular etiology of comorbidities in other complex diseases affecting disparate anatomical sites.
medRχiv
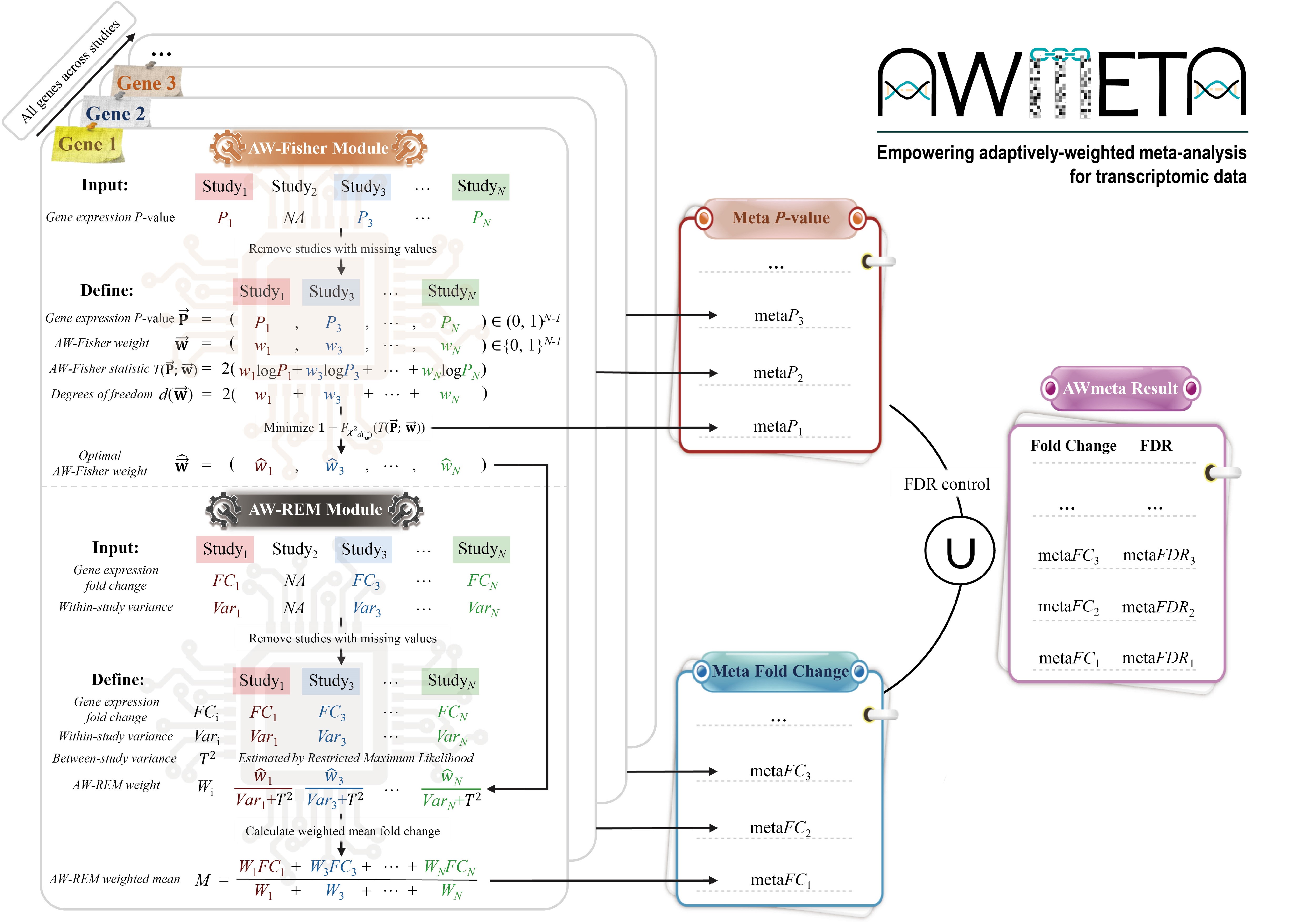
AWmeta empowers adaptively-weighted transcriptomic meta-analysis
Transcriptomic meta-analysis enhances biological veracity and reproducibility by integrating diverse studies, yet prevailing P-value or effect-size integration approaches exhibit limited power to resolve subtle signatures. We present AWmeta, an adaptively-weighted framework that unifies both paradigms. Benchmarking across 35 Parkinson's and Crohn's disease datasets spanning diverse tissues and adaptively down-weighting underpowered studies, AWmeta yields higher-fidelity differentially expressed genes (DEGs) with markedly reduced false positives and establishes superior gene differential quantification convergence at both gene and study levels over state-of-the-art random-effects model (REM) and original studies. AWmeta requires fewer samples and DEGs from original studies to achieve substantial gene differential estimates, lowering experimental costs. We demonstrate AWmeta's remarkable stability and robustness against external and internal perturbations. Crucially, AWmeta prioritizes disease tissue-specific mechanisms with higher functional coherence than those from REM and original studies. By bridging statistical rigor with mechanistic interpretability, AWmeta harmonizes heterogeneous transcriptomic data into actionable insights, serving as a transformative tool for precision transcriptomic integration.
bioRχiv
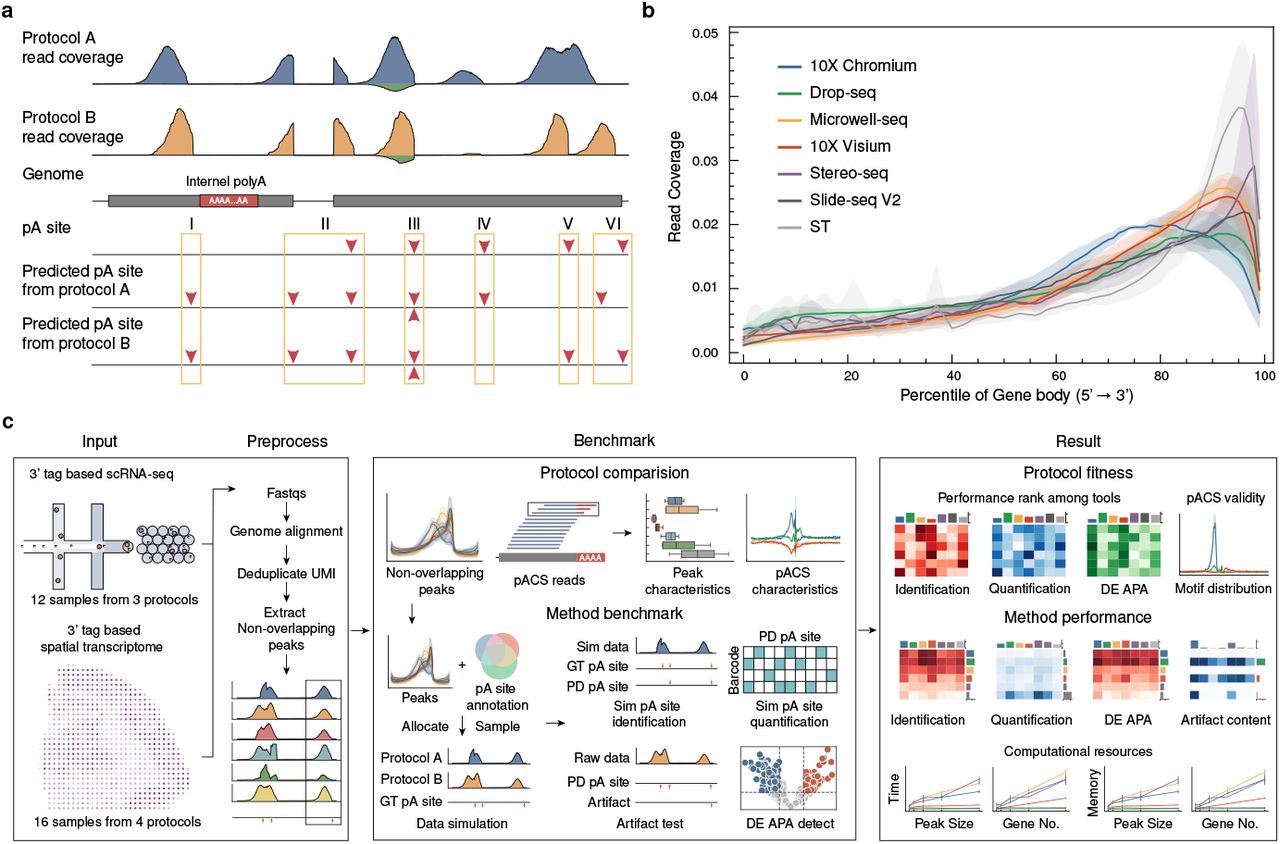
Benchmarking alternative polyadenylation detection in single-cell and spatial transcriptomes
3’-tag-based sequencing methods have become the predominant approach for single-cell and spatial transcriptomics, with some protocols proven effective in detecting alternative polyadenylation (APA). While numerous computational tools have been developed for APA detection from these sequencing data, the absence of comprehensive benchmarks and the diversity of sequencing protocols and tools make it challenging to select appropriate methods for APA analysis in these contexts. We systematically compared seven 3’-tag-based sequencing protocols and identified key peak features affecting APA detection performance. We developed a simulation pipeline that generates realistic datasets preserving protocol-specific characteristics. Using simulated and real data, we comprehensively assessed six computational tools for their ability to identify polyA sites, quantify polyA site expression, detect differentially expressed (DE) APA genes, filter sequencing artifacts, and their computational efficiency. We also investigated factors influencing APA detection. Our evaluation revealed that SCAPE and scAPAtrap generally outperformed other tools across various performance metrics and protocols. Our systematic evaluation provides guidance for tool selection, experiment design, and future tool development in APA analysis for single-cell and spatial transcriptomics, paving the way for investigating APA in these contexts.
bioRχiv
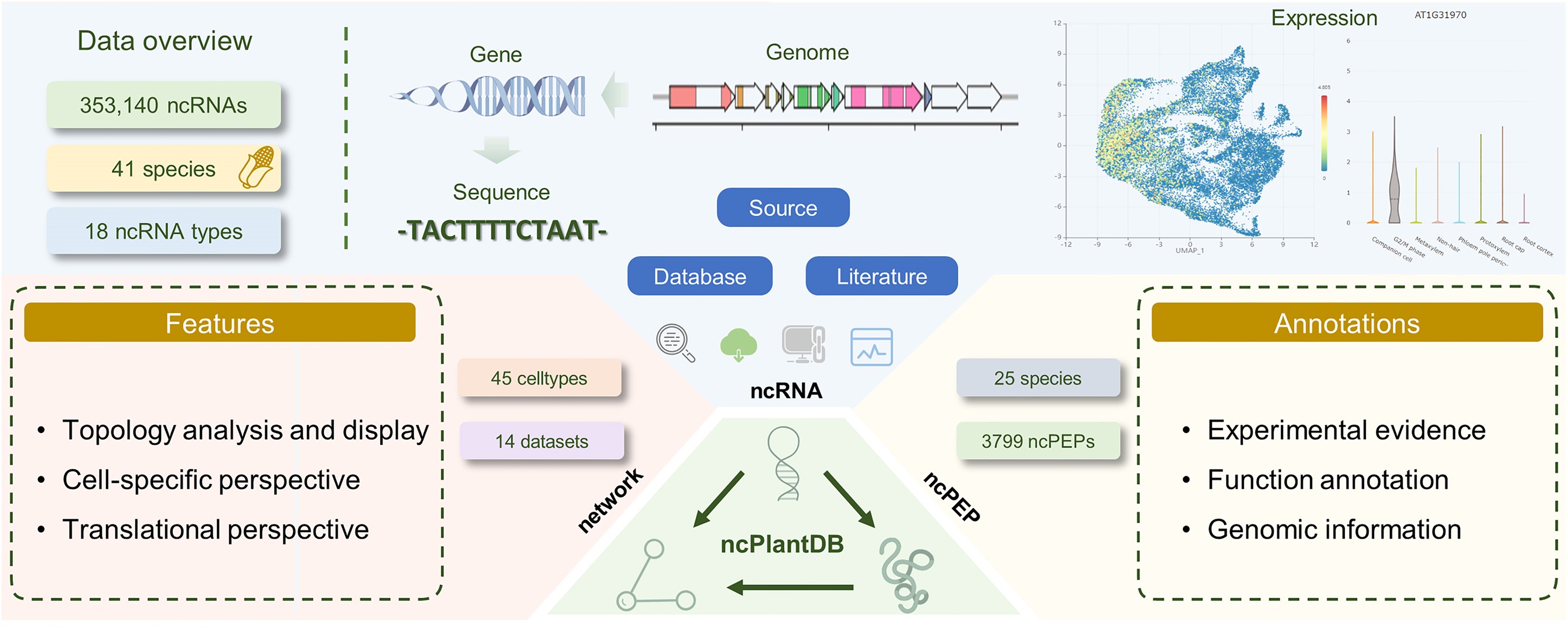
ncPlantDB: a plant ncRNA database with potential ncPEP information and cell type-specific interaction
The field of plant non-coding RNAs (ncRNAs) has seen significant advancements in recent years, with many ncRNAs recognized as important regulators of gene expression during plant development and stress responses. Moreover, the coding potential of these ncRNAs, giving rise to ncRNA-encoded peptides (ncPEPs), has emerged as an essential area of study. However, existing plant ncRNA databases lack comprehensive information on ncRNA-encoded peptides (ncPEPs) and cell type-specific interactions. To address this gap, we present ncPlantDB (https://bis.zju.edu.cn/ncPlantDB), a comprehensive database integrating ncRNA and ncPEP data across 43 plant species. ncPlantDB encompasses 353 140 ncRNAs, 3799 ncPEPs and 4 647 071 interactions, sourced from established databases and literature mining. The database offers unique features including translational potential data, cell-specific interaction networks derived from single-cell RNA sequencing and Ribo-seq analyses, and interactive visualization tools. ncPlantDB provides a user-friendly interface for exploring ncRNA expression patterns at the single-cell level, facilitating the discovery of tissue-specific ncRNAs and potential ncPEPs. By integrating diverse data types and offering advanced analytical tools, ncPlantDB serves as a valuable resource for researchers investigating plant ncRNA functions, interactions, and their potential coding capacity. This database significantly enhances our understanding of plant ncRNA biology and opens new avenues for exploring the complex regulatory networks in plant genomics.
Nucleic Acids Research
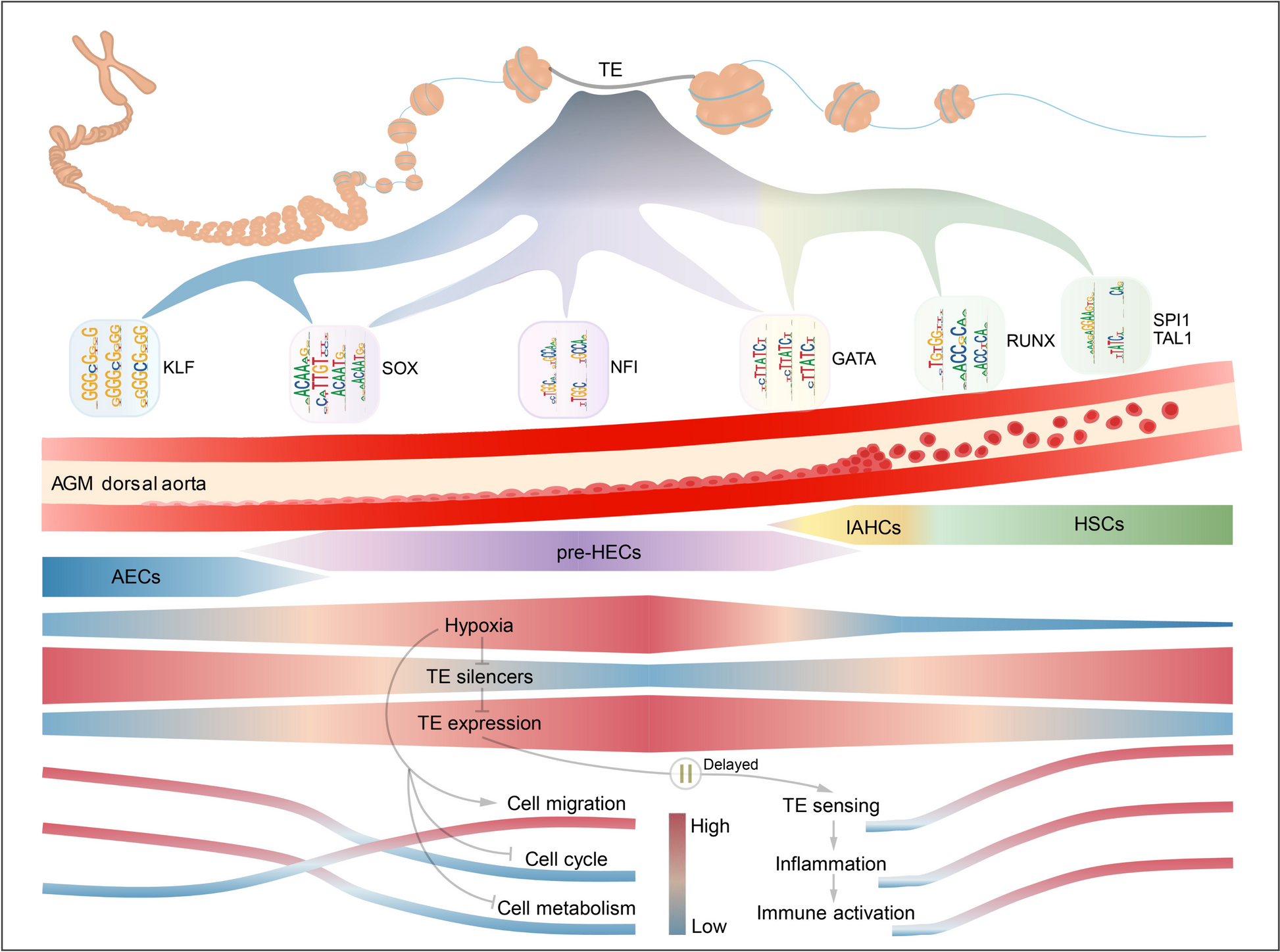
Systematic single-cell analysis reveals dynamic control of transposable element activity orchestrating the endothelial-to-hematopoietic transition
The endothelial-to-hematopoietic transition (EHT) process during definitive hematopoiesis is highly conserved in vertebrates. Stage-specific expression of transposable elements (TEs) has been detected during zebrafish EHT and may promote hematopoietic stem cell (HSC) formation by activating inflammatory signaling. However, little is known about how TEs contribute to the EHT process in human and mouse. We reconstructed the single-cell EHT trajectories of human and mouse and resolved the dynamic expression patterns of TEs during EHT. Most TEs presented a transient co-upregulation pattern along the conserved EHT trajectories, coinciding with the temporal relaxation of epigenetic silencing systems. TE products can be sensed by multiple pattern recognition receptors, triggering inflammatory signaling to facilitate HSC emergence. Interestingly, we observed that hypoxia-related signals were enriched in cells with higher TE expression. Furthermore, we constructed the hematopoietic cis-regulatory network of accessible TEs and identified potential TE-derived enhancers that may boost the expression of specific EHT marker genes. Our study provides a systematic vision of how TEs are dynamically controlled to promote the hematopoietic fate decisions through transcriptional and cis-regulatory networks, and pre-train the immunity of nascent HSCs.
BMC Biology
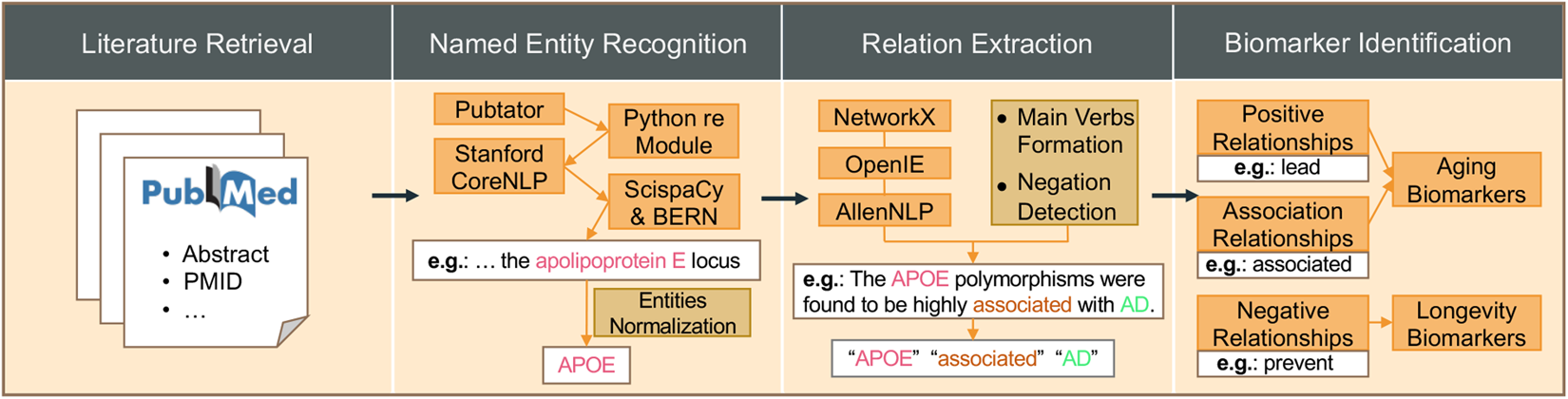
HALD, a human aging and longevity knowledge graph for precision gerontology and geroscience analyses
Human aging is a natural and inevitable biological process that leads to an increased risk of aging-related diseases. Developing anti-aging therapies for aging-related diseases requires a comprehensive understanding of the mechanisms and effects of aging and longevity from a multi-modal and multi-faceted perspective. However, most of the relevant knowledge is scattered in the biomedical literature, the volume of which reached 36 million in PubMed. Here, we presented HALD, a text mining-based human aging and longevity dataset of the biomedical knowledge graph from all published literature related to human aging and longevity in PubMed. HALD integrated multiple state-of-the-art natural language processing (NLP) techniques to improve the accuracy and coverage of the knowledge graph for precision gerontology and geroscience analyses. Up to September 2023, HALD had contained 12,227 entities in 10 types (gene, RNA, protein, carbohydrate, lipid, peptide, pharmaceutical preparations, toxin, mutation, and disease), 115,522 relations, 1,855 aging biomarkers, and 525 longevity biomarkers from 339,918 biomedical articles in PubMed. HALD is available at https://bis.zju.edu.cn/hald.
Scientific Data
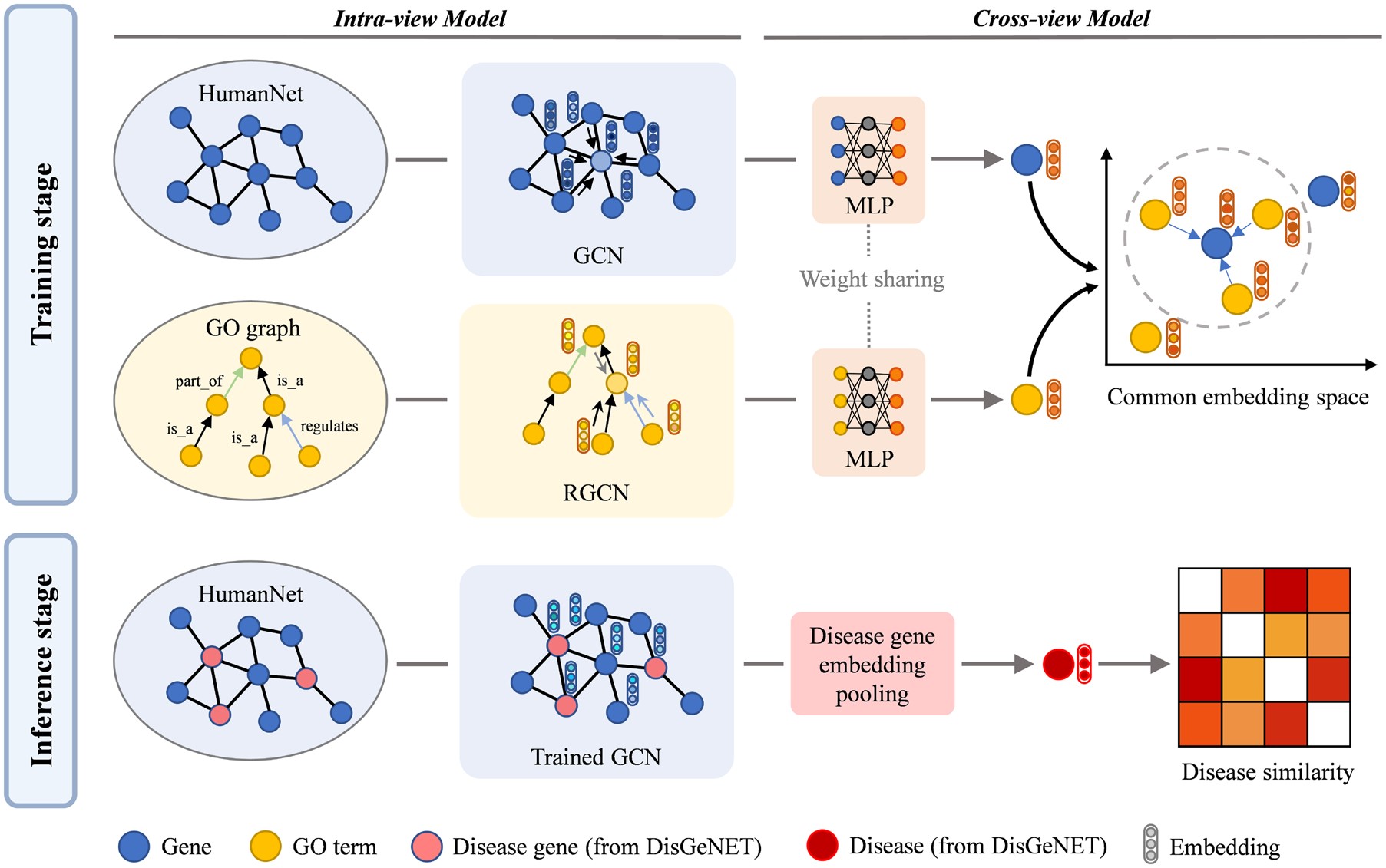
CoGO: a contrastive learning framework to predict disease similarity based on gene network and ontology structure
Quantifying the similarity of human diseases provides guiding insights to the discovery of micro-scope mechanisms from a macro scale. Previous work demonstrated that better performance can be gained by integrating multiview data sources or applying machine learning techniques. However, designing an efficient framework to extract and incorporate information from different biological data using deep learning models remains unexplored. We present CoGO, a Contrastive learning framework to predict disease similarity based on Gene network and Ontology structure, which incorporates the gene interaction network and gene ontology (GO) domain knowledge using graph deep learning models. First, graph deep learning models are applied to encode the features of genes and GO terms from separate graph structure data. Next, gene and GO features are projected to a common embedding space via a nonlinear projection. Then cross-view contrastive loss is applied to maximize the agreement of corresponding gene-GO associations and lead to meaningful gene representation. Finally, CoGO infers the similarity between diseases by the cosine similarity of disease representation vectors derived from related gene embedding. In our experiments, CoGO outperforms the most competitive baseline method on both AUROC and AUPRC, especially improves 19.57% in AUPRC (0.7733). The prediction results are significantly comparable with other disease similarity studies and thus highly credible. Furthermore, we conduct a detailed case study of top similar disease pairs which is demonstrated by other studies. Empirical results show that CoGO achieves powerful performance in disease similarity problem.
Bioinformatics


